Bioinformatics Tutorials by Santhilal :: RNA-seq
- RNA-seq analysis pipeline
- Introduction to RNA-seq
- Installation of tools
- Preparing annotations
- Introduction to formats
- Downloading RAW data
- Sample quality
- Cleaning reads
- Alignment
- Alignment quality
- Gene quantification
- Using R
- Differential expression
- Prepare Files for DE analysis
- edgeR analysis
- Annotate genes
- Saving the results
- Top candidates
- DE summary
- Enriched pathways
- Extra task
- RNA-seq analysis of lung cancer
Introduction to RNA sequencing
This session is a lecture. Lecture slides will be sent to the participants via email after the seminar.Tip for this tutorial
Before finishing one step do not look at next step to avoid confusion.RNA sequencing tools
Transcriptome is most implemented and analysed technique after microarray and whole genome sequencing. Therefore there are plenty of tools available to perform the analysis. The ideal way of choosing the right tools is by reading the articles on benchmarking tools for specific task of RNA-seq analysis. Tip: Always choose core bioinformatics journal to look for benchmarking article or any top journals which publish bioinformatics methods and protocols.Here is an example for recent benchmarking article for RNA-seq aligners,
"Simulation-based comprehensive benchmarking of RNA-seq aligners"
Question RS1: If you are given to choose between HISAT2 and STAR aligner, which one is your choice (based on the above article) ? Tip: Read the article, there is no need to understand the figures.
If you are too busy for searching article and reading, you can have a alternate option to ask bioinformaticians from the forums and blogs like, Biostars, seqanswers, BOL etc,. Biostars is the best one to get immediate reply.
Installing tools:
Recent days most of the tools developed does not require any special installation procedures. It is just simple as,1. Download
2. Extract/unzip/uncompress
3. Use the tool
For installing 'programs' I am creating 'bin' in home directory as I explained during lecture '/home/santhilal/bin'. Sorry I forgot to mention during lecture. Always good to have a common folder were you download and keep the tools or programs. In this tutorial we calll it as 'bin' in your home directory, which is to store all programs/tool you use for analysis.
$$ cd /home/santhilal/bin/ $$ wget http://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.5.zip $$ unzip fastqc_v0.11.5.zip
$$ cd /home/santhilal/bin/ $$ wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/downloads/hisat2-2.0.5-Linux_x86_64.zip $$ unzip hisat2-2.0.5-Linux_x86_64.zip
$$ cd /home/santhilal/bin/ $$ wget https://superb-dca2.dl.sourceforge.net/project/samstat/samstat-1.5.1.tar.gz $$ tar -zxvf samstat-1.5.1.tar.gz
$$ cd /home/santhilal/bin/ $$ wget https://excellmedia.dl.sourceforge.net/project/subread/subread-1.5.1/subread-1.5.1-Linux-x86_64.tar.gz $$ tar -zxvf subread-1.5.1-Linux-x86_64.tar.gz
$$ cd /home/santhilal/bin/ $$ wget https://github.com/samtools/samtools/releases/download/1.9/samtools-1.9.tar.bz2 $$ tar -xvjf samtools-1.9.tar.bz2 $$ cd samtools-1.9 $$ ./configure $$ make
Preparing/organizing directories and annotation files for the analysis
Preparing directories:
Always remember the most important step in bioinformatics analysis is to organize your folders well before designing your pipeline and starting your analysis. Please create the directories (folders) and sub-directories as shown in the following diagram. Example: I am creating the below directory structure in '/data1/santhilal/'. The 'programs' directory refers to the 'bin' directory we created in home '/home/santhilal/bin'.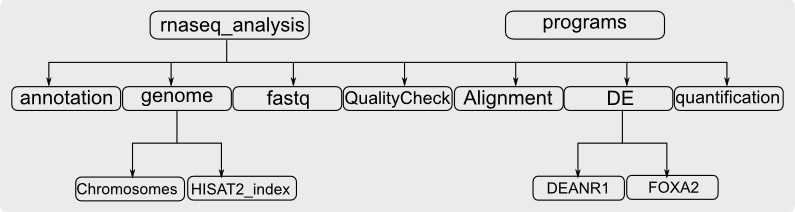
Prepare the annotation files for your organism:
In this step we are going to prepare the reference genome and reference transcript annotation. Reference genome is used for aligning the raw reads from sequencing.Question RS2: What is the purpose of reference transcript annotation ? Tip: Read the slides from the lecture.
Genome annotation (FASTA):
Example for FASTA format: Usually the extension of the file names may end with '.fasta' or '.fa'. The below mentioned sequence represents FASTA formatted data containing part of the sequence from Chromosome 1.
>chr1 CTGATGGTATGTACCTTTAATATAATTTGGTGAGAATGGTACTTCACCTC TGTGGTCTTCCTCCCCAAAACCCATAACCCCAGTCAAACCCTAAGAAAAA CATCAGCCAAACTCAAATTTAGGGCATTCTGCAAAAATAACTGACTGACT AGTACTCCCTCAAACTGTCAGGGTCACCAAAAACAAGGAAAGTCTAAGAA ACTCACAGCCAAGAGGTACCTAAAATGACAAATAAATTCAATGTGGTGGC CCGGATGGGATCCTGGAACAAAGAAAAAGAATGTTAGCTAAAAACTAAGG AAATCTAAATAAATGTATAGGCTTTAGTTAATAATACTCTATTGAATATT CATTAGTTGTGACAAATATGAGAGGTTAGTAATAGGAAAAAAATGGGAAC TCTGTGTATCTTTGAAATTTTTCTGTATATCTGAACTATTCTAAAATTTT AGTACCACTAAAGGTTTTTATTACATAATAGTGTTTTAATTTAATCTCCA TGCTATTTAATGAAATTTACCAGTTGTATTTGTGTTTGATTAGACATTAT CTCAAGTTGATTGGAATTACTCATATTTCTTAATGCCATTGTTGCAGCTG TAATAAGGTTGACTTTTCTACAATAGACACAGAAATCATTACATCACCAT TTAATTTTTGAAAAATAATCCGATCCCCTGTGATTATAAACTTGAGATTC TTTAAGAAAGTAGAAAGGTTGTAAGATATTATGAGTCATAGGATATTTCC AGTTTTGAAGTAATAAGACAAAAATCATTAACTAAACAAATTGGCATTAA AAAAAAACAGTATTTTTGAGAGGACCAGTCCAGACAATGTGTCTGTAAGA TAGACTTTACAAACTTAGTTTGATTAATTTTTGTTTTTCCGAGGATATGA TTCTTTTCTATAATTACTGTTCTGTTTTTTTCTGACAGGCTATCAAATAT TGGCTGCCTTAAGCTCTCTGTGAACTGAGTTATTCTCAATAATACTCTGT1) Download the recent version of genome file for Homo sapiens from Ensembl database in directory 'genome/Chromosomes'. Navigate to the directory and then type this command.
$$ cd /data1/santhilal/rnaseq_analysis/genome/Chromosomes $$ wget ftp://ftp.ensembl.org/pub/release-87/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gzNote: File downloaded/link obtained from source 'http://www.ensembl.org/info/data/ftp/index.html'
2) After downloading your files please decompress the '.fa.gz' files using (please make sure you are in directory '/data1/santhilal/rnaseq_analysis/genome/Chromosomes' and list the files after decompressing),
$$ gunzip -d Homo_sapiens.GRCh38.dna.primary_assembly.fa.gzTranscript annotation:
In this practical session we will be using transcript annotation from ensembl database. Download the GTF (Gene Transfer Format) file in directory '/data1/santhilal/rnaseq_analysis/annotation' and decompress it.
$$ wget ftp://ftp.ensembl.org/pub/release-87/gtf/homo_sapiens/Homo_sapiens.GRCh38.87.gtf.gzNote: File downloaded/link obtained from source 'http://www.ensembl.org/info/data/ftp/index.html'
RAW data and sequencing file formats
Keep in mind, it is always good to use compressed file before start your analysis to save some space on server or local (uncompressed file is 10-fold larger than compressed one). The sequencing platform usually provide RAW data in FASTQ format either compressed or un compressed. The uncompressed file will have extension on '.fastq' or '.fq' and compressed file will have '.fastq.gz' or '.fq.gz'. If you are getting uncompressed file, always good to compress it using following command (all RNA-seq aligners supports compressed form as input),$$ gzip sample.fastqThe above command will generate a compressed file 'sample.fastq.gz'. In this tutorial I will provide you with already compressed file. To view fastq file you can use 'cat' and for compressed fastq use 'zcat' (remember to display only head).
Example for FASTQ format:
@SRR1958169.1 HISEQ:269:HAMADADXX:1:1101:1168:1870 length=51 NGCGTGGAAGGGGTGGAGCCGCACCCGGATATGGAAGCCATCTTTGCCACA +SRR1958169.1 HISEQ:269:HAMADADXX:1:1101:1168:1870 length=51 #1=DDFFFHHHHHFHIIJJJJJJJJJJJJJJJJIJJJJJJHHHHHHFFFFF @SRR1958169.2 HISEQ:269:HAMADADXX:1:1101:1295:1887 length=51 NTAGGGTGAGCCCCACATCGGCCTGTGTATATCCCAGGGTGATCCTCTTCT +SRR1958169.2 HISEQ:269:HAMADADXX:1:1101:1295:1887 length=51 #1=DDDDDHHHHHIIIIIIDDGHGE9?:?DGDFHDD<BDH;FFHGGGIIIH
Downloading RAW dataset from GEO repository
Dataset will be used from published study "The lncRNA DEANR1 Facilitates Human Endoderm Differentiation by Activating FOXA2 Expression". Steps to be followed to download the RAW sequencing file from the GEO repository.
1) Go to https://www.ncbi.nlm.nih.gov/geo/. In the search box paste the GSE accession id/number you found from the article (Tip: You can easily find GEO id in PDF of the article).
2) Note down and copy the PRJNAxxx (project id) and get information about the samples from http://www.ebi.ac.uk/ena
3) In the 'Select Columns' section select (check the box) 'Experiment title and Sample title'.
4) You will find 'WT', 'FOXA2KD' and 'DEANR1KD' samples (2 replicate each = 6 samples) along with other samples from the study.
5) Save the generated text file by clicking on the link 'Text'. Save it with name 'PRJNA192215.txt'.
6) You will find the links in 'fastq_ftp' column of the text file. Download the samples corresponding to 'WT', 'FOXA2KD' and 'DEANR1KD' knockdown samples (6 samples).
7) Rename the files into simple names 'WT_1.fastq.gz', 'WT_2.fastq.gz', 'DEANR1KD_1.fastq.gz', 'DEANR1KD_2.fastq.gz', 'FOXA2KD_1.fastq.gz' and 'FOXA2KD_1.fastq.gz'.
Downloading: Below mentioned command will download the files from the path mentioned in the downloaded information file 'PRJNA192215.txt'. Since downloading will take long time, you can submit this job in background. In the following command I am redirecting normal nohup.out and saving into 'download_nohup.out'. If you do not want to rename you just use nohup YOUR_COMPLETE_COMMAND &. This will save your progress in default 'nohup.out'.
Tip: (nohup/background processing)
$$ cd /data1/santhilal/rnaseq_analysis/fastq/ $$ nohup cat /data1/santhilal/rnaseq_analysis/fastq/PRJNA192215.txt | sed -n '3,8p' | cut -f2 | xargs wget > download_nohup.out 2>&1 & # Print only 3rd to 8th row from the file 'PRJNA192215.txt' and cut column 12 containing download linksKeep checking tail/last lines of 'download_nohup.out'. If it is still downloading you will see different line each time you 'tail' nohup.out. Orelse simple relay on 'top' command. Easy and reliable way to check is 'grep "saved" /data1/santhilal/rnaseq_analysis/fastq/download_nohup.out'. When the sample is downloaded and saved to your disk, wget assigns for each sample 'saved' status. Once you see 'saved' six times means all your six samples completed downloading and saved to your disk.
Checking sequence quality
To check the quality of reads from the sequencing platform we will use 'FastQC' tool (popular). There are also plenty of other tools available for this purpose.$$ nohup /home/santhilal/bin/FastQC/fastqc -o /data1/santhilal/rnaseq_analysis/QualityCheck/ -t 6 /data1/santhilal/rnaseq_analysis/fastq/*.fq.gz &Remember from the above command you are running all 6 jobs parellel. You can utilize 'top' command and 'nohup.out' file to check the status of submitted job (Refer lecture slides).
Example for Good Illuminal data
Example for Bad Illuminal data
Remove sequence adapters or cleaning reads
Since the downloaded samples are already subjected to cleaning. We do not need this step. Most of the time the deposited sequences are free from adapters. Also sequencing facilities provide cleaned reads, in case you request. It is always better to request the sequening facility to clean the reads, so that we do not have to guess what kind of adapter sequence used by the facility.Aligning sequence to genome
In this step we will be aligning our sequencing reads to the genome. For this purpose, we use alignment/assembly tool. There are tools specifically designed for RNA sequencing. Because RNA-seq reads needs special handling of splice sites while mapping to genome. Always before starting the mapping, we need to index the genome according to the aligner's guideline. In this tutorial we are using HISAT2 aligner and it has its own indexing method. The first step here is to index the downloaded genome and next we are going to align using HISAT2. HISAT2 indexing: For indexing the input is our downloaded genome file and output should be saved to appropriate indexing directory. Below job submission was assigned with 8 threads (you can choose based on the availability). there will be multiple files generated with the name 'hg38*' in the indexing folder ('HISAT2_index').$$ cd /data1/santhilal/rnaseq_analysis/genome/HISAT2_index $$ nohup /home/santhilal/bin/hisat2-2.0.5/hisat2-build -p 8 /data1/santhilal/rnaseq_analysis/genome/Chromosomes/Homo_sapiens.GRCh38.dna.primary_assembly.fa /data1/santhilal/rnaseq_analysis/genome/HISAT2_index/hg38 &HISAT2 Alignment: Next step is alignment of downloaded fastq files to the indexed genome. Since we are going to submit more than one alignment jobs in the background. All the logs from the submitting jobs will be overwritten to the same file '/data1/santhilal/rnaseq_analysis/Alignment/nohup.out'. To avoid this, we are going to give unique 'nohup' name for each submission. For that we will be renaming the default 'nohup.out' to a name appropriate for the job. Every alignment program outputs alignment in SAM format.
Sequence Alignment or Map format (SAM): The compressed or binary format of SAM files is called BAM (Binary Alignment or Map) format. Always remember to output the alignment in BAM format because SAM format takes 10 times of more storage space compared to BAM. The HISAT2 alignment always produce SAM file format as output. We need to parse/pipe to output from HISAT2 to samtools and instantly converting it to BAM without saving the SAM file and finally sorting the BAM file by chromosome names. Sorting is necessary for some read quantification programs like HTSeq-count ut in this tutorial we use featureCounts for quantification which do not expect BAM file to be sorted.
$$ cd /data1/santhilal/rnaseq_analysis/Alignment $$ mkdir WT_1 WT_2 DEANR1KD_1 DEANR1KD_2 FOXA2KD_1 FOXA2KD_2 $$ nohup /home/santhilal/bin/hisat2-2.0.5/hisat2 --phred33 -p 8 --qc-filter -x /data1/santhilal/rnaseq_analysis/genome/HISAT2_index/hg38 -U /data1/santhilal/rnaseq_analysis/fastq/DEANR1KD_1.fastq.gz | /home/santhilal/bin/samtools-1.9/samtools view -@ 8 -bS - | /home/santhilal/bin/samtools-1.9/samtools sort -@ 8 -O BAM -o /data1/santhilal/rnaseq_analysis/Alignment/DEANR1KD_1/DEANR1KD_1_sorted.bam - &Similarly submit alignment jobs for other samples based on available CPUs/memory on the server.
Example of SAM file format:
SRR1958167.113755 256 10 27135 1 51M * 0 0 TCATCACCAAATAGTGCTACCTAAAGTAATATACAACCAATTTAATAAGTT CCCFFFFFHHHHHJHGIJJJJJJJJJHIJJJJJJJJJJJJJJJJJJJJJIJ NH:i:3 HI:i:2 AS:i:50 nM:i:0 SRR1958167.6172299 256 10 27763 1 51M * 0 0 AGCTCAGCCATGGCCTTGGGTCTGAAGCACAGAGCTGTGTGGAGTCTGGGC B@CFFFFFHHHHHJJJJJJJIJJJIJJJJJJHIIHIJJJIIIIJJJJJJJJ NH:i:3 HI:i:2 AS:i:50 nM:i:0 SRR1958167.16554448 256 10 28612 0 1S50M * 0 0 GGCAGGAGAGACTGAGGTGATGAGGGTCCAGAGTGGACCCCCAGAAAACCA CCCFFFFFHGHHHJIJJFHIIJJJIJIJJJJIJGIJJJIJJJIJIJIJJJJ NH:i:5 HI:i:5 AS:i:49 nM:i:0 SRR1958167.4929810 272 10 31436 0 51M * 0 0 GATCACCTGAGGTGAGGAGTTTGAGACCAGCCTGACCAACATGGAGAAACC HD999F;FDG?3IIGFHCIIIIIHHEHGHGECBEIIIIHGHHFEDDB=1@= NH:i:7 HI:i:4 AS:i:50 nM:i:0 SRR1958167.11547776 272 10 31436 0 51M * 0 0 GATCACCTGAGGTGAGGAGTTTGAGACCAGCCTGACCAACATGGAGAAACC JIIHHJJJJJJJJJIJJJJJJJJIIIJJJJIIJJIJJJHHHHGFFFFFBCC NH:i:7 HI:i:4 AS:i:50 nM:i:0 SRR1958167.23532591 272 10 31436 0 51M * 0 0 GATCACCTGAGGTGAGGAGTTTGAGACCAGCCTGACCAACATGGAGAAACC JJIIHJJJJJJJJJJJJJJJJJJJJIJJJJJJJJJJJJHHHHHFFFFDB@B NH:i:7 HI:i:4 AS:i:50 nM:i:0 SRR1958167.4156273 272 10 31546 0 51M * 0 0 AGCTACTTGGGAGGCTGAGGCAGGAGAGTCACTTGAACCCGGGAGGCAGAG IJGJJJJJJJIJJJJJJJJJJHJJIJJJJIIJJJJHGJHHHHHFFFFFCCC NH:i:10 HI:i:2 AS:i:50 nM:i:0 SRR1958167.19438259 16 10 31558 1 47M4S * 0 0 GGCTGAGGCAGGAGAGTCACTTGAACCCGGGAGGCAGAGGTTGCGGTGGGC HAEHGEGEGHGGCIIIEHFIJIIIJJIIIIJGGIHIJJHHHHHFFFDFCC@ NH:i:4 HI:i:3 AS:i:46 nM:i:0 SRR1958167.6675573 272 10 39012 0 51M * 0 0 AGACACAACCAAAAAAGAGAATTTTAGACCACTATCCTTGATGAACATTGG DBGHEIHFJJJJJJIHEJJHGGHHHHGHIIJJIJJJGIFHDHHFFDDB@@@ NH:i:10 HI:i:2 AS:i:48 nM:i:1 SRR1958167.6855956 0 10 46995 3 1S50M * 0 0 CGGCTGTCAGAACACAGTAAAGAATCCACACTGCTTCCCCCCTTTACCTAG CCCFFFFFHHHHHJIJJIJJJJIJJJJJJJJIJIJJJJJJJJHIJHJJIJG NH:i:2 HI:i:1 AS:i:49 nM:i:0Refer SAM format manual section 1.4 for complete description.
Check alignment quality
To check the alignment quality we are going to use SAMstat.$$ export PATH=$PATH:/home/santhilal/bin/samtools/samtools $$ /home/santhilal/bin/samstat-1.5.1/src/samstat /data1/santhilal/rnaseq_analysis/Alignment/DEANR1KD_1/DEANR1KD_1.bamor you can run all the jobs in background.
$$ export PATH=$PATH:/home/santhilal/bin/samtools-1.9/samtools $$ cd /data1/santhilal/rnaseq_analysis/Alignment $$ nohup /home/santhilal/bin/samstat-1.5.1/src/samstat /data1/santhilal/rnaseq_analysis/Alignment/*/*.bam > nohupSAMstat.out 2>&1The above command will generate a HTML file '/data1/santhilal/rnaseq_analysis/Alignment/DEANR1KD_1/DEANR1KD_1.bam.html'. This file can be opened with any web browser. More information on SAMstat is here.
Quantifying samples using transcript annotation
After aligning the samples and checking the alignment quality, we are going to quantify the aligned reads for our transcript annotation (GTF). Subread is a package provides read counting script called featureCounts. There are other popular tools like HTSeq tool for quantification. HTSeq is based on python and often prone to errors while installing because of python syntax keeps on changing between versions. Also there was question of reproducibility by using HTSeq. Although recent versions has solved this issue and HTSeq gives almost same output as featureCounts. Some problems faced by HTSeq users is 1) installation problems, 2) need to keep track of python versions, 3) job execution is lot slower than featureCounts, and 4) no support for parallel jobs/multiple threads. Subread featureCounts does not need installation, less error prone, can run parallel jobs and it is super-fast.For quantification, we need to provide GTF files containing information about genes (by transcripts and exons) and our BAM files. This will outputa a table containing reads from individual samples gene-wise in the order we provide. Examples/guideline, running featureCounts. Complete USER GUIDE for featureCount quantification is on page 30 of user guide.
$$ nohup /home/santhilal/bin/subread-1.5.0-p1-source/bin/featureCounts -T 6 -t exon -g gene_id -Q 30 -F GTF -a /data1/santhilal/rnaseq_analysis/annotation/Homo_sapiens.GRCh38.87.gtf -o /data1/santhilal/rnaseq_analysis/quantification/hg38_ensembl_GRCh38.87_counts.txt /data1/santhilal/rnaseq_analysis/Alignment/WT_1/WT_1_sorted.bam /data1/santhilal/rnaseq_analysis/Alignment/WT_2/WT_2_sorted.bam /data1/santhilal/rnaseq_analysis/Alignment/DEANR1KD_1/DEANR1KD_1_sorted.bam /data1/santhilal/rnaseq_analysis/Alignment/DEANR1KD_2/DEANR1KD_2_sorted.bam /data1/santhilal/rnaseq_analysis/Alignment/FOXA2KD_1/FOXA2KD_1_sorted.bam /data1/santhilal/rnaseq_analysis/Alignment/FOXA2KD_2/FOXA2KD_2_sorted.bam > nohupFeatureCounts.out 2>&1
Differential Expression analysis
There are plenty of R Bioconductor tools available for differential expression (DE) analysis (eg., edgeR, DESeq2, DEGSeq, EBSeq etc.,). Here we are going to use classic DE tool edgeR.Preparing files for DE
Prepare design matrix: Prepare a plain text file with,1) First column (samples) containing infromation about the column/samples names from the quantification file we generated using featureCounts.
2) Next column (labels) in design matrix can be optional or just have some extra infromation.
3) Third column (condition) containing name of sample groups. Example, 'WT_1' and 'WT_2' we are going to use as one group called 'control'. This column is very important because you are going to tell edgeR that which samples belong to which group.
samples labels condition WT_1 control1 control WT_2 control2 control DEANR1KD_1 DEANR1KD1 DEANR1 DEANR1KD_2 DEANR1KD2 DEANR1 FOXA2KD_1 FOXA2KD1 FOXA2 FOXA2KD_2 FOXA2KD2 FOXA2Prepare quantification table: From the quantification table, we will have lot of information which will not be required in next steps. We have to remove those extra columns and just create a matrix with the required information.
$$ cat /data1/santhilal/rnaseq_analysis/quantification/hg38_ensembl_GRCh38.87_counts.txt | grep -v "^#" | cut -f1,7,8,9,10,11,12 | sed '1d' | sed '1i\Geneid\tWT1_1\tWT1_2\tDEANR1KD_1\tDEANR1KD_2\tFOXA2KD_1\tFOXA2KD_2' > /data1/santhilal/rnaseq_analysis/quantification/hg38_ensembl_GRCh38.87_counts.matrixPrepare annotation table: Make a gene-wise table from downloaded GTF ('/data1/santhilal/rnaseq_analysis/annotation/Homo_sapiens.GRCh38.87.gtf') as explained in the Main assignment 2 and save it to the directory '/data1/santhilal/rnaseq_analysis/annotation/' with name 'Homo_sapiens.GRCh38.87_gene_annotation.txt'.
edgeR analysis
Now we are ready for performing differential expression analysis. To start with, we first need to start R session from your terminal and, install and load edgeR package as shown below. Since edgeR is an bioconductor package, we need to follow installation instructions from bioconductor repository.Instruction: You need to just copy paste the command between '<<' and '#'. Example form the below command, the portion maked in red is the actual command you have to copy and paste it in your R terminal. The text after '#' is just description of the command for your understanding.
$$ R
## try http:// if https:// URLs are not supported
>> source("https://bioconductor.org/biocLite.R") # Connecting repository
>> biocLite("edgeR") # Installing
>> library("edgeR") # Loading installed edgeR libraries
Load your design matrix to a variable 'mydesign' and your quantification table in 'x' variable. Remember we prepared an annotation table using GTF, load that into 'dbs' variable. Final thing is specifying the path where your outputs from edgeR to be saved (in this case the 'outpath' will have path stored in the variable). In the following commands, - header=T means, header is TRUE and it takes first line from the file as header/table-header. Alternate representation is header=TRUE.
- sep="\t" means, in the file each columns are separated with TAB delimitation.
- row.names=1 means, considering first column as rownames (Require to mention for edgeR analysis).
>> mydesign <- read.table("/data1/santhilal/rnaseq_analysis/DE/exp_design.txt", header=T, sep="\t")
>> x <- read.table("./data1/santhilal/rnaseq_analysis/quantification/hg38_ensembl_GRCh38.87_counts.matrix", header=T, sep="\t", row.names=1)
>> dbs <- read.table("/data1/santhilal/rnaseq_analysis/annotation/Homo_sapiens.GRCh38.87_gene_annotation.txt", header=T, sep="\t")
>> outpath <- "/data1/santhilal/rnaseq_analysis/DE/"
Check if the above variables are created properly by using 'ls()'. Next check the contents of the variables just by typing the variables names you created above (if the variable has huge data, do not print everything instead you can just print first ten entries using head).
>> ls() >> mydesign >> head(x) >> head(dbs) >> outpathGive information about the groups to edgeR along with loaded counts matrix.
>> group <- factor(mydesign$condition) >> d <- DGEList(counts=x, group=group)Normalizing reads to counts per million (CPM) and filtering the genes by keeping only the genes with CPM > 5 in atleast one sample (prominantly expressed in atleast one sample). We should be carefull in choosing this because if you are analyzing for non-coding RNAs, this value (cpm>5) is too high. Since our aim in this exercise is to get only top significantly expressed protein-coding genes, we are using more stringent criteria.
>> dim(d) >> keep <- rowSums(cpm(d)>5) >= 1 >> d <- d[keep,] >> dim(d) >> d$samples$lib.size <- colSums(d$counts)Dispersion estimates between samples (even within group) and between groups. This estimate is important for edgeR to calculate the differential expression because it gives information about how close or how variable the samples or groups. Read more about dispersion on page 44 of edgeR user guide.
>> d <- calcNormFactors(d) >> d1 <- estimateCommonDisp(d, verbose=T) >> d1 <- estimateTagwiseDisp(d1)## Plotting correlation of samples with normalized counts
>> png(paste0(outpath,"sample_correlation.png")) >> pheatmap(cor(log2(cpm(d)+1)), main="SampleCorrelation") >> dev.off()## Plotting library sizes of samples (Total number of sequenced reads assigned to all genes from the GTF file)
>> png(paste0(outpath,"sample_librarySizes.png")) >> barplot(d$samples$lib.size,las=2,names=colnames(d),cex.names=0.6,horiz=T,main="LibrarySizes") >> dev.off()## Differential expression between WT and DEANR1
>> et <- exactTest(d1, pair=c("control","DEANR1")) # Testing for differential expression betwee groups
>> res <- topTags(et, n=length(et)) # Extracting differentially expressed genes (if you just specify, topTags(et) will give you first ten DEGs)
>> res$table[,"Geneid"] <- rownames(res$table) # making rownames of the results to be first column of the results with column name 'Geneid' (easy for saving)
>> colnames(res$table) # Checking the columns of results
>> resF <- res$table[,c("Geneid","logFC","logCPM","PValue","FDR")] # Storing selective columns to a variable 'resF'
Annotating significant genes
We are now ready to merge our 'dbs' with information from GTF and DE results. The resulting table will have all information about genes from GTF with staus of differential expression (statistics generated by edgeR).>> annotated <- merge( dbs, resF, by='Geneid')
Saving DE results
## Saving complete DE analysis without any filtering. Usually the DE analysis is filtered based on 'FDR' and 'logFC' cutoff.>> write.table(annotated,paste0(outpath,"DEANR1/DEANR1_DEGs_annotated.txt"), sep="\t", quote=F,row.names=F)## Saving normalized read counts (counts per million) for all samples
>> nc <- cpm(d1$counts, normalized.lib.sizes=FALSE) # Normalization
>> nc <- cbind(rownames(nc),nc) # converting rownames ('Geneid') into first column of 'nc', it will be useful for annotation.
>> colnames(nc)[1] <- "Geneid" # Naming the first column as 'Geneid'
>> nc_annotated <- merge( dbs, nc, by='Geneid') # Merging 'dbs' and 'nc' by matching column 'Geneid'
>> write.table(nc_annotated,paste0(outpath,"normalized_cpm.txt"), sep="\t", quote=F,row.names=FALSE)
Filtering significant gene list
Now we try to filter the differentially expressed genes using log2fold change >= 1 and FDR<=0.05. In case of log2fold change we also need to get negative fold change values (downregulated genes). To achieve this we are going to tell R to convert the values in column 'annotated$logFC' to absolute values (abs) and then filter the genes having >=1 absolute log2fold change. In this way you will get all up- and downregulated genes.Note: In simple term absolute value means converting all values to positive, abs(-1)=1 and abs(+1)=1. # Filtered with log-fold change >=1 and FDR < 0.05 (all classes of genes and transcripts)
>> annotated_filt <- subset(annotated,abs(annotated$logFC)>=1 & annotated$FDR<=0.01) >> write.table(annotated_filt,paste0(outpath,"DEANR1/DEANR1_DEGs_annotated_filtered_all.txt"), sep="\t", quote=F,row.names=F)# Filtered with log-fold change >=1 amd FDR < 0.05 (only protein coding)
>> annotated_filt_pc <- subset(annotated,abs(annotated$logFC)>=1 & annotated$FDR<=0.01 & annotated$Class=="protein_coding") >> write.table(annotated_filt_pc,paste0(outpath,"DEANR1/DEANR1_DEGs_annotated_filtered_PC.txt"), sep="\t", quote=F,row.names=F)
Differential Expression summary
>> summary(annotated_filt$Class)Question RS3: Find differentially expressed protein coding genes between WT and FOXA2. Submit the complete edgeR code for this analysis.
Question RS4: How many protein coding genes are differentially expressed in DEANR1 and FOXA2 ? How many protein coding genes are common between them ? Do you get all the genes highlighted in Figure 5F from this study?
Question RS5: Report the missing genes from Figure 5F and your analysis. Please also report the status of missing gene (log-fold change and FDR of atleast one) and speculate on why are those genes missing from our analysis?
Functional/pathway enrichment analysis
Follow @GeneSCFVote on: Biostars
Question RS6: Perform geneontology (GO) biological processes (BP) enrichment analysis using GeneSCF tool for,
1) Protein coding DEGs in DEANR1
2) Protein coding DEGs in FOXA2
3) Common protein coding genes between DEANR1 and FOXA2
What are the top significantly GO_BP in all the above analysis?
Tip: Main assignment 2 Part 1
Differential expression analysis between lung cancer (smokers) patients and normal lung tissue samples from non-smokers.
1) Use RNA-seq samples form this publication.3) Select 5 samples from each group (5 cancer + 5 normal tissues).