Bioinformatics Tutorials by Santhilal :: UNIX
- Basic UNIX Commands
- Introduction
- wget
- ls
- pwd
- mkdir
- cp/rm/mv
- cd
- head/more/less
- cat
- wc
- grep
- unique
- sort
- comm
- Advanced pipe
- sed
- tr
- join
- Advanced UNIX
- awk
- Assignments
- BASH scripting
Getting Started
Introduction
In order to practice UNIX commands you need a workspace called shell or shell prompt. This shell is an interface which passes commands to the UNIX system to do different tasks depend on the commands provided by the user. All the commands from this tutorial can be run on Linux/Ubuntu terminal or Microsoft Windows 10 bash utility. Get description of commands from http://explainshell.com.Retrieve files or directories from online (FTP/HTTP)
Since you do not have any files in your home directory, you will be using the files prepared for this course. With wget utility from unix, you can download any files from web to your local directory on Linux.$$ wget http://kandurilab.org/users/santhilal/courses/UNIX/materials/course_file_repo.zipEvery command in unix has its own help section. To check options available for individual commands you can use '--help' or '-h' after the command to explore the help section or documentation. Test this with 'wget' command by typing 'wget --help' (long format argument) or 'wget -h' (short format argument). Note: You will also learn in this course how to retrieve multiple files from the web at once in later part of the tutorial.
Listing files in the directory/folder
List the files and folders (contents) in your home folder.Shows all the files and folders in the current directory:
$$ ls course_file_repo.zipShows all the files and folders in the current directory in long listing format:
$$ ls -l -rw-rw-r-- 1 santhilal santhilal 1538218 Mar 9 15:05 course_file_repo.zip
$$ ls -lh -rw-rw-r-- 1 santhilal santhilal 1.5M Mar 9 15:05 course_file_repo.zipQuestion U1: What is the difference between 2nd and 3rd command ? In what situation 3rd command will be useful ?
Tip: Read more information on ls command from the following link.
Check your current working directory/folder
$$ pwd /home/santhilal
Create directory/folder
$$ mkdir practice $$ ls course_file_repo.zip practice
Copy / Remove/ Rename/ Create folder/files
$$ cp course_file_repo.zip practice/ $$ ls course_file_repo.zip practiceAbove command copied your file to the newly created directory 'practice'. Now you have two copies of 'course_file_repo.zip', one in home directory and other in 'practice' directory. You remove the one from your home directory to manage your space efficiently. Whenever you remove some file, always check whether you are on the right directory or removing the right file. Because in command line you do not have 'undo' option to bring your files back.
TIP: Whenever you want to move some files to other directory, I always recommend use copy 'cp' command to copy from source to destination directory and then use 'rm' to remove it from the source directory. You may use the directly the move 'mv' command to move your files from source to destination directory at your own risk.
$$ pwd $$ /home/santhilal $$ rm course_file_repo.zip $$ ls $$ practiceTask: We are going to,
1) Create a folder named 'dummy' and some files inside the folder 'dummy' named 'file1.txt' and 'file2.txt'
2) Edit the content of the files
3) Rename folder to 'mydummy'
4) Rename files to 'file1.txt' -> 'myfile1.txt', 'file2.txt' -> 'myfile2.txt'
5) Remove file from the directory
6) Remove a complete folder
Step 1:
$$ mkdir dummy $$ cd dummy $$ nano file1.txt file2.txtThe above 'nano' command will create two files and open two files as empty files. For switching between files use 'ALT+.' (Next file) and 'ALT+,' (Previous file). Now switch to 'file1.txt' and type "This is my first file" and save the file using 'CNTRL+O' (it will ask to confirm the file name, press ENTER to confirm). Now you saved 'file1.txt' and to exit the 'file1.txt' press 'CNTRL+X'. The 'file1.txt' will exit and show the remaining file which is 'file2.txt'. Type "This is my second file", save and exit the file.
Step 2: Add second lines to both the files as 'First file is now edited' ('file1.txt') and 'Second file is now edited' ('file2.txt')
$$ nano file1.txt file2.txtStep 3:
$$ pwd $$ /home/santhilal/dummy $$ cd .. # Since you cannot be inside the directory and rename the same directory you are in, we will go back one directory by using this command. $$ mv dummy mydummyStep 4:
Similarly please rename 'file1.txt' -> 'myfile1.txt', 'file2.txt' -> 'myfile2.txt'
Now let's merge the created text files using command 'cat' (concatenate).
$$ cat myfile1 myfile2 > mergedfile.txt # This merges the contents of the file in a new file $$ cat mergedfile.txt # Display the content of merged fileStep 5: Removing/deleting file
$$ rm mergedfile.txtRemoving files start with 'myfile'
$$ rm myfile* # Removes any files start with name 'myfile'. Here '*' means match anything after 'myfile'. This is called pattern matching.Step 6: To remove directory and their sub-directories or files inside the directories, we command the shell to 'recursively (-r)' remove all the contents.
$$ rm -r mydummy
Change or navigate directories. Now we are going to navigate to the directory where we stored our downloaded course material.
$$ cd practice/ $$ ls course_file_repo.zipNow uncompress the zip file using 'unzip' command. If unzip command is not found/installed, please install by typing.
$$ sudo apt-get install unzipUnzip using following command.
$$ unzip course_file_repo.zipQuestion U2: How many folders are in 'course_file_repo.zip' ? Please check the size of individual files in each folder and report.
Display part of the contents of the file
Now navigate to directory/folder 'gene_lists'.$$ cd gene_listsList the files from the directory.
$$ lsTry these commands.
$$ head H0.list $$ head H0.list H12.list $$ less H0.list $$ more H0.list Note: To exit from 'more' and 'less' commands, just type 'q'.
Display complete text file
$$ cat ambiguous.listWARNING: If the file is bigger (say million lines) and you executed the above command, it will start printing files line by line on the screen. To stop printing just type 'Cntrl+c'.
Count words or lines
$$ wc -l H0.listTo get number of genes from all the list in the directory.
$$ wc -l *.listIn the above command '*' is wild-card character. It tells the command to check number of genes/lines in the files where the file names ends with '.list' (match all the files ending with '.list'). Try commands 'wc' and 'wc -c' over files.
Pattern matching using grep
In this section you will be introduced to the concept of piping, where you pass output of one command to the next command. You can do unlimited number of piping in unix. Now we are going to match word or pattern in the files (pipe: combining different commands to get required output) Match genes start with 'USP'.$$ cat H0.list | grep "^USP*" # Here '^' means 'line starts with' $$ cat H0.list | grep "USP*"Match genes ending with '.' followed by some digit/number.
$$ cat H0.list | grep "\.[0-9]$"From the above command you will see all the genes ending with some number with preceeding one character/number match. And in the above command '\' is used to escape '.' character not to be used as pattern matching expression. See the difference by removing '\'. As you know from previous section that '*' matches with any number of letter/character/digit. In the above command we are using '.', which in pattern matching means match anythin but only one letter/character/digit. That is the reason we have to tell 'grep' command to consider '.' as character but not as pattern matching entity. Here '[0-9]' means any number ranging from 0 to 9. And '$' represents the ending part of pattern matching. If you have anything after number, it will not match since you wanted the gene name to end with number. Lean more about pattern matching from the following link. Try these examples to know the significance of '.' in pattern matching.
$$ cat H0.list | grep "USP." $$ cat H0.list | grep "USP.."The first command will match one character after 'USP' and the next one matches two character after it.
Question U3: How do you print the number of genes starts with 'RP11-' and ends with number '3' in H0.list and H12.list? I want you to print the number, not the genes. Please write the command you used for getting the results.
Get unique items from the file
Check what are the genes repeated in the list 'ambiguous.list' (duplicates),$$ cat ambiguous.list | sort | uniq -dCheck genes without duplicate entries (unique),
$$ cat ambiguous.list | sort | uniq -uMaking a list without any duplicate gene entries. By default 'uniq' command gives you the unique entries from the file (even without using -u argument). Tip: you save any commands output just by using '>' (redirection) followed by new/output filename.
$$ cat ambiguous.list | sort | uniq > ambiguous_noduplicates.list
Sorting items in file
$$ sort H0.list > H0_sorted.list $$ sort H12.list > H12_sorted.listQuestion U4: In the above example, you will get sorted list as 'A->Z'. What will be your command to get 'Z->A' order ?
Number of overlapping entries between two lists
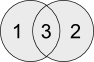
Overapped genes between H0 and H12 gene lists (suppress unique genes from 1 and 2)
$$ comm -12 H0_sorted.list H12_sorted.list | wc -lGenes unique to H0 (suppress unique genes from 2 and 3)
$$ comm -23 H0_sorted.list H12_sorted.list | wc -lGenes unique to H12 (suppress unique genes from 1 and 3)
$$ comm -13 H0_sorted.list H12_sorted.list | wc -lQuestion U5: I would like you to save the files from all the above commands as 'H0_H12_overlapped.list', 'H0_specific.list' and 'H12_specific.list'. Write your command ?
Multiple/advanced piping
$$ cd annotation $$ nano hg38_gene_annotation_mart_export.txtWhenever you open a file or dataset, observe the structure of the dataset well. Now we are going to count number of genes, transcripts or isoforms in the annotation file. Also we are going to check how many protein coding genes are in the annotation. Most of the genes has multiple isoforms. We are using human annotation from ensembl database in this tutorial. In ensembl annotation, the gene names always start with 'ENSG' and isoforms/transcripts of those gene names starts with 'ENST'. Below image shows the structure of the file 'hg38_gene_annotation_mart_export.txt' where you can find SERTAD4-AS1 has gene name 'ENSG00000280963' and it has two transcripts variants or isoforms such as 'ENST00000625817' and 'ENST00000628975'. Similarly you can also see that 'FOXO6' also has two isoforms.
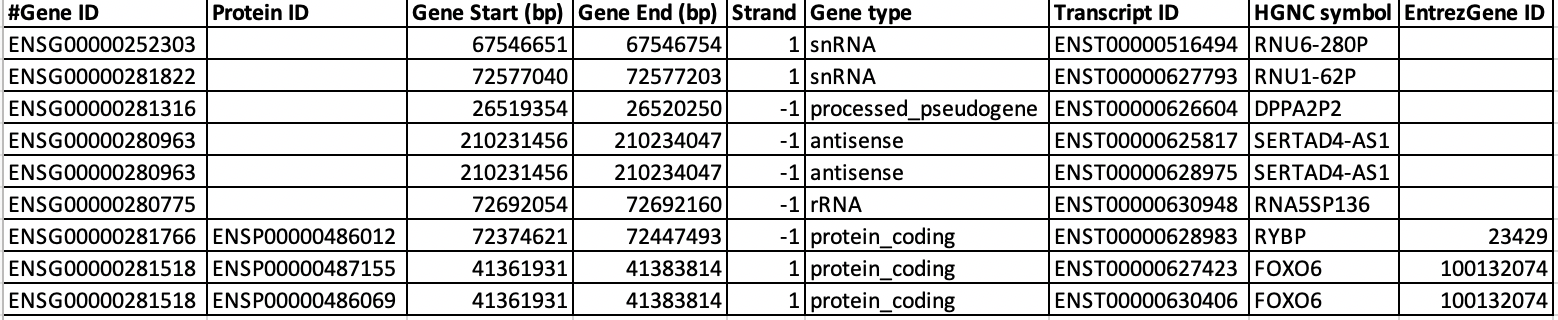
$$ cat hg38_gene_annotation_mart_export.txt | grep -v "^#" | cut -f1 | sort | uniq | wc -l # This counts total number of unique genes in the annotation $$ cat hg38_gene_annotation_mart_export.txt | grep -v "^#" | cut -f7 | sort | uniq | wc -l # This counts total number of unique transcripts $$ cat hg38_gene_annotation_mart_export.txt | grep -v "^#" | grep -w "protein_coding" | cut -f1 | sort | uniq | wc -l # Counts only total number of protein coding genesQuestion U6: How many isoforms are there for gene 'TRAPPC4' ?
Question U7: How many different Gene type (exampe, protein_coding, antisense etc.,) are there in the annotation file ?
Stream editor used for replacing/inserting/deleting content of file
'Sed' command is one of the border/bridge between basic and advanced unix commands. This command has lot of functionality. We are going to learn only limited functionality in this course (this is enough for working with the RNA-seq data). In this section we are going to make unstructured table into well structured table using only 'sed' command.Tidy Data A: Each line follows same format. Such as, column1_header<SPACE>"ID"<TAB>column2_header<SPACE>"NAME"... so on. But this format is not suitable for a table. Here we are going to convert this tidy (well-defined data: all lines follows same format) into proper table format as in second table (Pretty Table B).
gene_id "ENSG00000223972" gene_name "DDX11L1" chr1:11869-14409 gene_biotype "transcribed_unprocessed_pseudogene" + 2540 gene_id "ENSG00000227232" gene_name "WASH7P" chr1:14404-29570 gene_biotype "unprocessed_pseudogene" - 15166 gene_id "ENSG00000278267" gene_name "MIR6859-1" chr1:17369-17436 gene_biotype "miRNA" - 67 gene_id "ENSG00000243485" gene_name "MIR1302-2" chr1:29554-31109 gene_biotype "lincRNA" + 1555 gene_id "ENSG00000237613" gene_name "FAM138A" chr1:34554-36081 gene_biotype "lincRNA" - 1527 gene_id "ENSG00000268020" gene_name "OR4G4P" chr1:52473-53312 gene_biotype "unprocessed_pseudogene" + 839 gene_id "ENSG00000240361" gene_name "OR4G11P" chr1:62948-63887 gene_biotype "unprocessed_pseudogene" + 939 gene_id "ENSG00000186092" gene_name "OR4F5" chr1:69091-70008 gene_biotype "protein_coding" + 917 gene_id "ENSG00000238009" gene_name "RP11-34P13.7" chr1:89295-133723 gene_biotype "lincRNA" - 44428 gene_id "ENSG00000239945" gene_name "RP11-34P13.8" chr1:89551-91105 gene_biotype "lincRNA" - 1554Pretty Table B:
Geneid GeneSymbol Chromosome Class Strand Length ENSG00000223972 DDX11L1 chr1:11869-14409 transcribed_unprocessed_pseudogene + 2540 ENSG00000227232 WASH7P chr1:14404-29570 unprocessed_pseudogene - 15166 ENSG00000278267 MIR6859-1 chr1:17369-17436 miRNA - 67 ENSG00000243485 MIR1302-2 chr1:29554-31109 lincRNA + 1555 ENSG00000237613 FAM138A chr1:34554-36081 lincRNA - 1527 ENSG00000268020 OR4G4P chr1:52473-53312 unprocessed_pseudogene + 839 ENSG00000240361 OR4G11P chr1:62948-63887 unprocessed_pseudogene + 939 ENSG00000186092 OR4F5 chr1:69091-70008 protein_coding + 917 ENSG00000238009 RP11-34P13.7 chr1:89295-133723 lincRNA - 44428
We normally use 'cat' command to display the contents of the file or to pipe/pass the contents of the file to next command. In Unix, we also have an option to work with compressed file without uncompressing it (only works if it is compressed in '.gz' format). In this case we use 'zcat' instead of 'cat' command. You will find 'Homo_sapiens.GRCh38.87_badStructure.table.gz' file in the directory 'annotation'. Try to use 'head' after each pipe, to check how the output looks like from the previous command.
$$ zcat Homo_sapiens.GRCh38.87_badStructure.table.gz | sed 's/gene_id "//' | sed 's/gene_name "//' | sed 's/gene_biotype "//' | sed 's/"//g' | sed '1i\Geneid\tGeneSymbol\tChromosome\tClass\tStrand\tLength' | sed 's/ //g' | headPlease save the pretty Table B with file name 'Homo_sapiens.GRCh38.87_annotation.table'. Tip: you save any commands output just by using '>' (redirection) followed by new/output filename.
To become master in sed follow this documentation. Minimum knowledge about 'sed' (replacing, deleting, printing, inserting) is enough to deal with RNA-seq data analysis. For printing specific range of lines from the file.
$$ sed -n 1,2p Homo_sapiens.GRCh38.87_annotation.table $$ sed -n 4,6p Homo_sapiens.GRCh38.87_annotation.tableFor deleting lines
$$ cat Homo_sapiens.GRCh38.87_annotation.table | sed '1d' | headQuestion U8: Report the difference when you use "sed '1d' Homo_sapiens.GRCh38.87_annotation.table" and "sed -i '1d' Homo_sapiens.GRCh38.87_annotation.table" ?
Translate or replace content of file
Translate 'tr' command is a simple utility and not used extensively. Since there are better alternatives like 'sed', this command is less used. Only advantage of using 'tr' over other commands is when you want to translate a text from Lowe case to upper case or vice versa. Please practice 'tr' command from following link.Sample usage: The command 'echo' is used for printing anything on terminal. In this following case we are printing 'santhilal' and with the pipe passing it to translate 'tr' command. And 'tr' translates lower-case letters to upper-case letters.
$$ echo "santhilal" | tr a-z A-Z
Merge two files with specified column
You will find some list of genes which are differentially expressed in some experiment or analysis. We are going to annotate or add more information to those gene list using our file 'Homo_sapiens.GRCh38.87_annotation.table' which contains gene level annotation.Two step process: (Some times this will not work on Windows Bash)
$$ sort -k1 annotation/Homo_sapiens.GRCh38.87_annotation.table > annotation/Homo_sapiens.GRCh38.87_annotation_sorted.table $$ sort -k1 DEG_results/DiffExpressed_gene.list > DEG_results/DiffExpressed_gene_sorted.list $$ join annotation/Homo_sapiens.GRCh38.87_annotation_sorted.table DEG_results/DiffExpressed_gene_sorted.list -t $'\t' > DEG_results/DiffExpressed_gene.list.annotatedOne step process:
$$ join <(sort annotation/Homo_sapiens.GRCh38.87_annotation.table) <(sort DEG_results/DiffExpressed_gene.list) -t $'\t' > DEG_results/DiffExpressed_gene.list.annotatedInsert the column header:
$$ cat DEG_results/DiffExpressed_gene.list.annotated | head # Check the contents of annotated file $$ sed -i '1i\Geneid\tGeneSymbol\tChromosome\tClass\tStrand\tLength' DEG_results/DiffExpressed_gene.list.annotated # insert required column headers
Advanced UNIX
AWK
AWK is a different programming language developed by three programmers. It is mainly created to manipulate text files. In this course we will learn limited functionality of AWK.Changing the order of columns in a table: In this example we are going to change the order of columns from file 'DEG_results/DiffExpressed_gene.list.annotated' to 'Geneid\tGeneSymbol\tClass\tStrand\tChromosome\tLength'.
$$ cat DEG_results/DiffExpressed_gene.list.annotated | head
$$ cat DEG_results/DiffExpressed_gene.list.annotated | awk 'BEGIN{FS="\t"}{print $1"\t"$2"\t"$4"\t"$5"\t"$3"\t"$6}' | head
$$ cat DEG_results/DiffExpressed_gene.list.annotated | awk 'BEGIN{FS="\t"}{print $1"\t"$2"\t"$4"\t"$5"\t"$3"\t"$6}' > DEG_results/DiffExpressed_gene.list.annotated.ordered
Using if-condition, single condition::
Get the genes with length less than 200 nucleotides.
$$ cat DEG_results/DiffExpressed_gene.list.annotated.ordered | awk 'BEGIN{FS="\t"}{if($6<201) print $0;}' # In this case 'print $0' means, print all the columns. If you just want selected columns, you can specify as 'print $1,$3'.
Question U9: How many differentially expressed genes has length less than 200 nts ?Using if-condition, multiple condition: Get only DE protein coding and lincRNAs. In IF-condition, We use '==' for numbers and '~' (tilde) to match a string/character. The following command uses '||' (OR) which means, print the lines where third-column is 'protein_coding' OR 'lincRNAs'. Since single '|' means pipe in unix, AWK uses '||'.
$$ cat DEG_results/DiffExpressed_gene.list.annotated.ordered | awk 'BEGIN{FS="\t"}{if($3~"protein_coding" || $3~"lincRNA") print $0;}'
Check more operators here.Question U10: How do you get genes with length ranges from 100 to 150 nts ? How many genes are there with that range of lengths ?
Main Assignment 1
I would like you to use the complete annotation table file from 'annotation/Homo_sapiens.GRCh38.87_annotation.table' and convert it to a BED format using the commands from your previos lessons.Your table format: TABLE
Geneid GeneSymbol Chromosome Class Strand Length ENSG00000223972 DDX11L1 chr1:11869-14409 transcribed_unprocessed_pseudogene + 2540 ENSG00000227232 WASH7P chr1:14404-29570 unprocessed_pseudogene - 15166 ENSG00000278267 MIR6859-1 chr1:17369-17436 miRNA - 67 ENSG00000243485 MIR1302-2 chr1:29554-31109 lincRNA + 1555 ENSG00000237613 FAM138A chr1:34554-36081 lincRNA - 1527 ENSG00000268020 OR4G4P chr1:52473-53312 unprocessed_pseudogene + 839 ENSG00000240361 OR4G11P chr1:62948-63887 unprocessed_pseudogene + 939 ENSG00000186092 OR4F5 chr1:69091-70008 protein_coding + 917 ENSG00000238009 RP11-34P13.7 chr1:89295-133723 lincRNA - 44428Desired output or file: BED (BED file is used for marking and visulaizing the location of genes or genomic features on the genome browser). Learn more about BED file format from this link.
track name="ensembl_hg38_annotation" chr1 11869 14409 ENSG00000223972:DDX11L1 0 + chr1 14404 29570 ENSG00000227232:WASH7P 0 - chr1 17369 17436 ENSG00000278267:MIR6859-1 0 - chr1 29554 31109 ENSG00000243485:MIR1302-2 0 + chr1 34554 36081 ENSG00000237613:FAM138A 0 - chr1 52473 53312 ENSG00000268020:OR4G4P 0 + chr1 62948 63887 ENSG00000240361:OR4G11P 0 + chr1 69091 70008 ENSG00000186092:OR4F5 0 + chr1 89295 133723 ENSG00000238009:RP11-34P13.7 0 -
Tip: Make use of grep, awk and sed.